

{kind=link}
{kind=link}
Angelman综合征合并外斜视一例
[叶海昀, 冯金彩, 王斯颖, 李苑, 乔彤 ]
]
]
|
|


患儿, 女, 2岁7个月, 足月顺产, 出生时胎膜早破, 胎心音弱, 哭声弱, 无抢救病史。18个月时仅会扶站, 现仅会扶走, 无意识地发出“ 妈” 的音节, 愉快表情, 神智清, 认知差, 头发棕色, 双足外翻。映光法检查示双眼呈外斜视状态(可交替性注视), 交替遮盖示眼球由外向内动。三棱镜检查示-40△ , 眼球各方位运动可, 视力检查欠合作。验光示右-1.00-2.75× 10, 左+0.50DS/-2.75DC× 2, 双眼角膜透明, 虹膜色淡(见图1), 视网膜结构正常。听力检查正常。脑电图(Electroencephalogram, EEG)示δ 、θ 波在枕区波幅增加明显。MRI检查示脑内未见明显异常。患儿血样检测示染色体15q11 PWS/AS相关区域SNRPN、TUBGCP5、UBE3A、MKRN3、GABRB3、ATP10A、MAGEL2、NDN和 NIPA基因杂合缺失; SNRPN-in01a、SNRPN-E1、SNRPN-in01b、SNRPN-promoter和NDNa区域低甲基化, 表明该样本为母源性缺失。父母均未检测到15q11 PWS/AS相关区域拷贝数变异及甲基化异常。临床诊断为Angelman综合征合并外斜视。患儿以外斜视收住眼科, 于全身麻醉下行双眼外直肌后退术, 术后眼球正位, 各方向运动可(见图2)。
 | 图1. Angelman综合征合并外斜视患儿术前外观 患儿愉快表情, 头发棕色, 双眼虹膜色淡, 角膜映光示双眼呈外斜视状态(可交替性注视), 交替遮盖示眼球由外向内动, 三棱镜检查示外斜40△ Figure 1. Pre-operative appearance of patient with Angelman syndromecomplicated by exotropia. The patient presented with a happy disposition, brown hair, brown iris, and exotropia (alternative). The prism examination showed -40△ . |
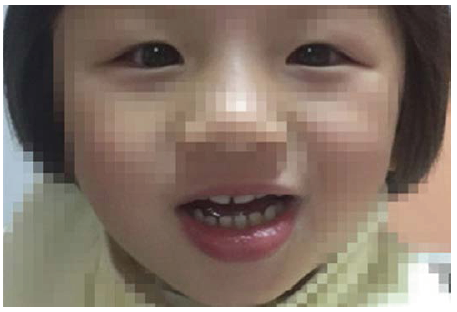 | 图2. Angelman综合征合并外斜视患儿斜视术后第1天外观 结膜轻度充血, 角膜映光位正, 交替遮盖无隐动Figure 2. Post-operative appearance of the patient with Angelman syndrome. There was mild conjunctival congestion, correct corneal light reflection, and the alternative cover test was normal. |
讨论:
Angelman综合征(Angelman syndrome, AS)是一种神经发育障碍性疾病, 其主要的临床表现为愉快面容、智力低下、运动或平衡障碍、严重语言障碍、行为异常及以巨大下颌及张口吐舌为特征的特殊面容等, 发病率约为1/15 000[1], 患者生活质量低下, 给家庭及社会造成极大负担。
1965年AS以“ 快乐木偶病” (Happy puppet disease)被认识, 其后陆续有文献报道。2006年Williams修订了AS的诊断标准[2]。AS和PraderWilli综合征(PraderWilli syndrome, PWS)均主要由染色体15q11区拷贝数变异和印记基因异常所导致, 父源性缺失或者母源性单亲二倍体导致PWS, 而母源性缺失或者父源性单亲二倍体将导致AS。PWS/AS综合征的缺失区域位于15号染色体上同一区域的P基因, 而该区域异常会导致眼皮肤白化病的发生(酪氨酸酶无关型)[2, 3, 4, 5, 6], 这可能与本病例头发棕色, 虹膜色淡的体征相关。目前AS主要有4种已知的遗传缺陷:15q11.2-13 区域的母源性缺失、父源性单亲二倍体(UPD)、印记缺损以及UBE基因(Ubiquitin-protein ligase, UBE3A)点突变, 不同基因型缺陷的患者临床表现存在差异[4, 5]。UBE3A蛋白广泛分布于脑组织细胞中, 以细胞核内最为富集, 其作为甾醇类激素受体的共同激活因子, 能联合不同转录因子继而结合DNA转录调控区, 参与下游基因转录调控并影响神经元的发育及可塑性[7]。AS的各种遗传机制均存在UBE3A的基因表达失败, 最常见的是15q11-13区段染色体微缺失, 见于70%~75%的患者, 多数缺失为母源性15q11-13缺失[5], 本例患儿即为此型, 其父母基因均正常。
AS患者常见的眼部表现包括虹膜色素减退, 脉络膜色素减退, 内斜视或外斜视, 屈光不正(通常远视)[4]; 其他表现还包括眼球震颤, 瞳孔反应迟钝, 视神经萎缩, 视网膜色素上皮细胞异常等。据研究报道, 斜视发生率为27%~ 75%, 可因基因缺失、UPD以及突变等变异而不同, 其中, 外斜视最为常见(9%~13%)[4]。本组病例即属于此类, 患儿眼外肌张力正常, 眼球各方位运动正常, 故手术时参考共同性外斜视手术的常规量进行手术设计, 术后眼位正位。
国内外研究人员总结了AS患儿特异性的EEG结果:如大量高波幅的δ 节律(2~3 Hz)或θ 节律(4~6 Hz), 长程发放或接近持续发放, 在慢波中夹杂棘波或尖波从而形成棘慢波或尖慢波图形等[6, 8], 这对AS的诊断具有高度敏感性, 然而EEG变异较大, 仍需要通过AS特征性的临床表现及基因检测才能最后确诊。
遗传学认为UBE3A基因的父母拷贝在其他组织中都是等同活性的, 仅在神经系统中父源性UBE3A基因被沉默, 母源性UBE3A基因正常表达[6, 9]。在AS患者神经元中, 父源性UBE3A基因沉默的同时, 母源性UBE3A基因发生突变。近期研究发现拓扑异构酶抑制剂可以阻断抑制父源性UBE3A基因表达的反义非编码RNA的产生, 研究者对缺失母源性UBE3A基因的小鼠进行静脉注射和脑内直接定点注射, 发现被沉默的父源性UBE3A基因功能被有效激活, 而且产生的UBE3A蛋白具有正常功能[9]。但目前是否可以用拓扑异构酶抑制剂来缓解人类各种AS的症状, 以及减小药物的毒性并增强药物的有效性都在进一步的研究中[6, 9]。
作者贡献声明 叶海昀:收集数据, 参与选题、设计及资料的分析和解释; 撰写论文; 对编辑部的修改意见进行修改。乔彤、冯金彩:收集病例, 参与选题、设计和修改论文的结果、结论。王斯颖、李苑:参与选题、设计、资料的分析和解释, 修改论文中关键性结果结论。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|