


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
先天性无虹膜家系中发现 PAX6基因新的致病突变
[邢怡桥1 , 李印1 , 李拓1  , 李家璋
, 李家璋2 , 杨琳1 ]
, 李家璋|
|


目的 对中国一先天性无虹膜家系进行 PAX6基因突变检测,以确定其突变位点。方法 实验研究。收集一先天性无虹膜家系,采集该家系患者及健康成员的外周静脉血,收集100名健康人外周血作为正常对照,采用Sanger测序的方法对 PAX6基因的11个外显子(外显子4-14)以及外显子-内含子连接区域进行测序,随后进行家系共分离分析以及正常样本的对照分析。结果 该家系8名成员经全面眼科检查,3名确诊为先天性无虹膜,且合并有复杂的眼部表型,包括不同程度的角膜病变、不同类型的白内障、黄斑发育不良、轻度上睑下垂和轻度眼球水平震颤等。在该家系患者中发现一个新杂合突变[c.569_570delinsACGG(p.Ile190Asnfs*18)],该突变可致 PAX6基因编码的蛋白截短,该突变符合共分离且在100名正常对照者中未检测到。结论 PAX6基因第8外显子上一个新的杂合突变[c.569_570delinsACGG(p.Ile190Asnfs*18)]为本研究中先天性无虹膜家系的致病突变,该突变与先天性无虹膜有关,本研究扩大了 PAX6基因的突变频谱。
Objective To identify the pathogenic cause of familial congenital aniridia by mutation screening of the paired box 6 ( PAX6) gene.Methods All participants in this experimental study, including the available family members of the recruited family and 100 unrelated healthy controls, received comprehensive ophthalmic examinations. Genomic DNA was extracted from peripheral blood. Mutation screening of 11 coding exons (exon 4 through exon 14) of PAX6 and the adjacent splicing junctions was performed by Sanger sequencing. Co-segregation analysis for the available family members and the normal controls were conducted later.Results A novel heterozygous deletion/insertion mutation c.569_570delinsACGG (p.Ile190Asnfs*18) in exon 8 of PAX6 was identified in the congenital aniridia family. This mutation consistently co-segregated with the affected family members and was not detected in the normal family members or in the 100 normal controls. Three in eight family members were diagnosed with congenital aniridia by comprehensive eye examinations. The patients with aniridia also had complications with heterogenic ocular manifestations, including keratopathy, different types of cataracts, macular dysplasia, mild ptosis, and mild horizontal nystagmus.Conclusion A novel PAX6 deletion/insertion mutation c.569_570delinsACGG (p.Ile190Asnfs*18) in exon 8 was the pathogenic mutation of the family and was associated with congenital aniridia. The finding expands the mutation spectrum of PAX6 in congenital aniridia.
先天性无虹膜是一种临床上较罕见且双眼发病的眼部疾病, 在不同人群中其发病率为1/96 000~ 1/64 000, 该疾病的主要特征是全部或部分无虹膜或虹膜发育不全, 通常还伴有其他眼部异常如角膜病变、白内障、青光眼、黄斑发育不良、斜视、眼球震颤和低视力等[1, 2, 3]。约2/3的先天性无虹膜病例为家族性, 多呈常染色体显性遗传, 具有较高外显率, 但表型各异, 其余1/3病例呈散发性, 无种族和性别差异[4]。
80%的先天性无虹膜患者与PAX6基因(OMIM 607108)突变有关[5], 该基因属于同源盒基因家族, 定位于人类11号染色体短臂1区3带(11p13), 包含14个外显子, 编码一个含422个氨基酸转录调控因子[6]。大部分的PAX6基因突变发生在外显子5-14上, 对于其中的少部分病例, PAX6基因突变可导致严重的表型异常。不同的突变可导致不同的表型, 然而, 相同的突变也可导致不同的表型[7, 8]。本研究中, 我们检测了一个先天性无虹膜家系的PAX6基因。该家系患者有着复杂的眼部表型:先天性无虹膜、角膜病变、白内障、眼球震颤、黄斑发育异常、轻度上睑下垂, 而且不同患者眼部表型严重程度不一。通过对PAX6基因的检测, 我们在第8外显子发现新的杂合突变位点[c.569_570delinsACGG(p.Ile190Asnfs* 18)], 包含2个碱基的缺失和4个碱基的插入, 该突变导致PAX6基因编码蛋白的截短而致病。
选择2016年9月在武汉大学恩施临床学院眼科中心检查确诊的一先天性无虹膜家系, 进行详细病史询问并确定该患者有家族史, 该家系3代共8名成员, 包括3名患者和5名正常人, 以上家系成员否认近亲婚配史。随机抽取100名与该家系无血缘关系的健康人群作为对照。上述3名患者均行肾脏多普勒彩超检查排除Wilms′ 瘤, 未发现全身其他系统并发症。家系图谱见图1。
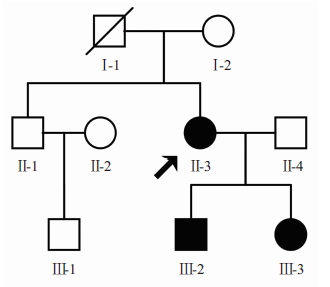 | 图1 先天性无虹膜家系图谱。方框代表男性, 圆圈代表女性, 黑色箭头指向先证者, 斜线代表该成员已去世。Figure 1 Congenital aniridia family diagram. Squares, males; circles, females; black arrow, proband case; slash, indicates the member has died. |
1.2.1 遗传学调查及样本采集 由经验丰富的眼科专家对该家系8名成员进行详细病史询问及全面眼科检查, 包括视力(裸眼视力和最佳矫正视力)、角膜、虹膜、晶状体、眼底、眼压。对所有患者进行眼前节及眼底照相存档(Ⅱ -3先证者由于角膜混浊无法行眼底检查), 行超声生物显微镜(UBM, SW-3200s, 天津索维公司)检查虹膜根部有无残留, 肾脏多普勒彩超检查排除Wilms瘤。对Ⅲ -3行黄斑OCT(Heidelberg Spectralis, 德国海德堡公司)检查了解黄斑发育情况(Ⅱ -3由于角膜混浊无法行黄斑OCT检查, Ⅲ -2由于眼球震颤及高度近视黄斑OCT检查结果不清晰)。该研究经过武汉大学恩施临床学院医学伦理委员会同意并遵守赫尔辛基宣言, 参与者均签署知情同意书。分别采集该家系8名成员以及100名健康对照者的外周静脉血, 均为5 ml, EDTA抗凝。
1.2.2 基因组DNA的提取及测序反应 采用经典酚-氯仿提取法提取所有研究对象外周静脉血白细胞基因组DNA, 引物设计参考文献[9], 由上海生工生物工程股份有限公司合成。所有反应均在GeneAmp PCR-9700(ABI, Foster City, CA)型温度梯度PCR仪上进行, 以稀释至40 ng/μ l的基因组DNA为模板, PCR反应体系为:Premix Taq(含2× GC Buffer、dNTPs及rTaq酶) 11.9 μ l, 上游引物0.5 μ l, 下游引物0.5 μ l, 模板DNA 1.6 μ l, 灭菌蒸馏水5.5 μ l, 共20 μ l。PCR反应条件:95 ℃预变性10 min; 按95 ℃变性30 s, 根据不同引物的退火温度退火复性30 s, 72 ℃延伸30 s的顺序, 循环35 次; 最后72 ℃延伸10 min。PCR扩增产物经Millipore纯化板纯化, 对纯化后的PCR产物加入BigDye做测序反应, 测序产物再次纯化, 纯化产物溶解后在ABI 3130 xl基因测序仪上对其进行双向直接测序, 测序结果采用DNAStar软件进行分析。
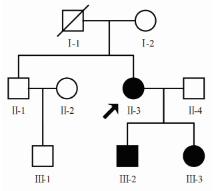
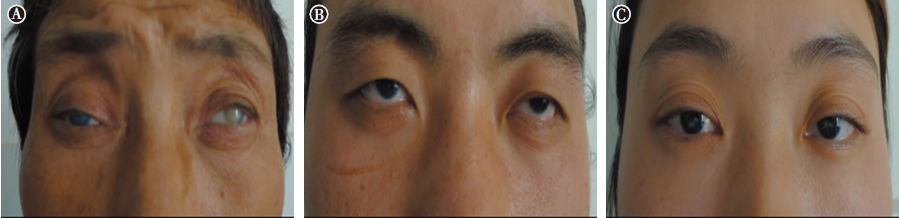 | 图2 Ⅱ -3(A), Ⅲ -2(B), Ⅲ -3(C) 3位先天性无虹膜患者眼表外观均显示轻度上睑下垂Figure 2 Ocular surface manifestation showed mild ptosis in three congenital aniridia patients (Ⅱ -3) A, (Ⅲ -2) B, and (Ⅲ -3) C. |
先证者(Ⅱ -3):女性, 56岁, VOD:眼前指数, VOS:眼前手动, 眼球水平震颤, 轻度上睑下垂(见图2A), 角膜全层混浊(见图3A-B), 右眼人工晶状体眼(2007年行右眼白内障超声乳化吸除术+人工晶状体植入术), 左眼晶状体窥不清, UBM提示虹膜缺如(资料未提供), 双眼底窥不清, 眼压正常。
先证者儿子(Ⅲ -2):男性, 28岁, 未婚, VOD:0.02, -15.5-1.75× 12° =0.08, VOS:0.02, -15.5-2.5× 101° =0.06, 双眼球轻度水平震颤, 轻度上睑下垂(见图2B)。角膜周边血管翳, 虹膜全部缺如, 晶状体点状混浊(见图3C-D), 眼底呈豹纹状改变, 眼压正常。
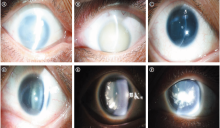
先证者女儿(Ⅲ -3):女性, 26岁, 未婚, VOD:0.1, -0.75-2.0× 10° =0.15, VOS:0.12(矫正不提高)。双眼球轻度水平震颤, 轻度上睑下垂(见图2C), 角膜周边血管翳, 虹膜全部缺如, 晶状体后囊下皮质混浊(见图3E-F), UBM检查可见虹膜缺如, 仅根部残留(见图4A), 眼底隐约可见(见图4B), 眼压正常。黄斑OCT检查右眼可见中心凹平坦, 提示黄斑发育不良(见图4C)。
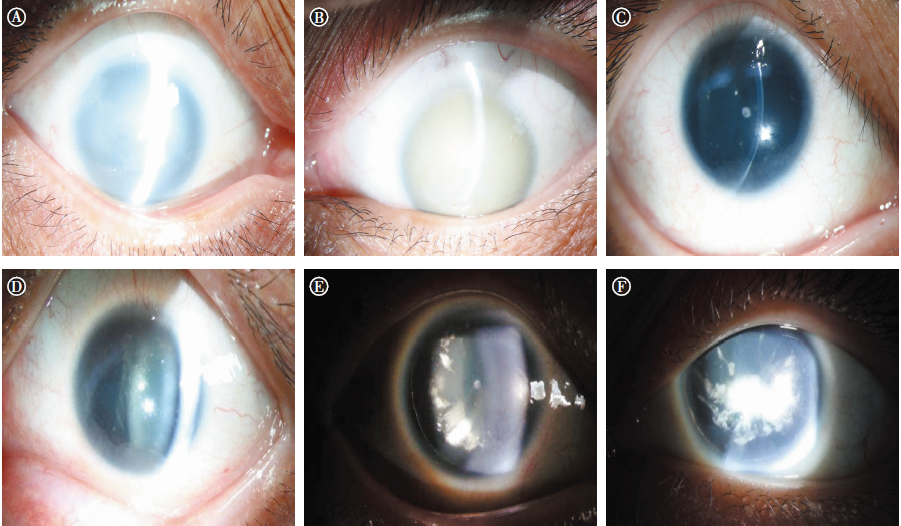 | 图3 3位先天性无虹膜患者眼前节照片。A:Ⅱ -3右眼角膜混浊, 血管翳生长, 隐约见虹膜缺如; B:Ⅱ -3左眼角膜全层混浊, 呈瓷白色, 虹膜不可见, 但UBM检查可见虹膜缺如; C:Ⅲ -2右眼角膜周边血管翳, 虹膜缺如; D:Ⅲ -2左眼角膜周边血管翳及周边混浊, 虹膜缺如; E:Ⅲ -3右眼虹膜缺如, 晶状体后囊下周边混浊; F:Ⅲ -3左眼虹膜缺如, 晶状体后囊下片状混浊区较右眼大Figure 3 Anterior segment photographs of the three congenital aniridia patients. A: Patient Ⅱ -3 had corneal opacity and pannus in the right eye, making the absence of the iris difficult to observe; B: Patient Ⅱ -3 had a serious corneal opacity of the left eye, the aniridia was detected by UBM; C: Patient Ⅲ -2 had pannus surrounding the right corneal limbus and aniridia; D: Patient Ⅲ -2 also had peripheral pannus and corneal opacity of the left eye and aniridia; E: Patient Ⅲ -3 had aniridia and posterior lamellar lens opacity in the right eye; F: Patient Ⅲ -3 had aniridia in the left eye and a poster lamellar lens opacity that was larger than in the right eye. UBM, ultrasound biomicroscopy. |
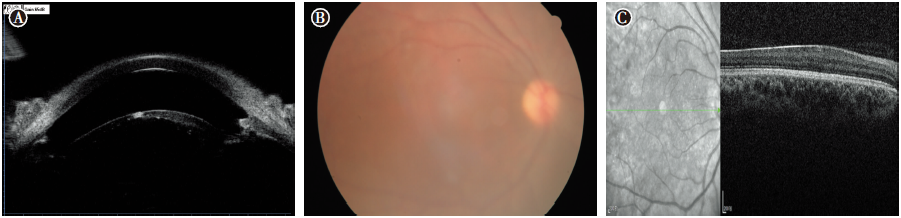 | 图4 先天性无虹膜患者Ⅲ -3的右眼UBM、眼底照相及黄斑OCT检查。A:UBM见少许虹膜根部残留; B:眼底照相示晶状体混浊, 眼底像模糊; C:黄斑OCT示中心凹平坦, 提示黄斑发育不良Figure 4 Ultrasound biomicroscopy, fundus photography, and optical coherence tomography of the right eye of congenital aniridia patient Ⅲ -3. A: UBM showed only a small residual portion of the iris in the right eye; B: Fundus photography of the right eye was blurry due to crystalline turbidity; C: OCT showed a flat off center fovea, suggesting macular dysplasia. UBM, ultrasound biomicroscopy; OCT, optical coherence tomography. |
对所有研究对象的PAX6基因进行双向测序, 结合GenBank基因组数据库, 采用DNAStar软件对测序结果进行对比分析发现, 该家系3名患者(Ⅱ -3, Ⅲ -2, Ⅲ -3)的PAX6基因第8外显子均存在杂合突变c.569_570delinsACGG, 即同时存在2个碱基的缺失合并4个碱基的插入, 见图5。该突变经网站软件Mutalyzer(http://www.lovd.nl/mutalyzer/)预测, 可产生氨基酸序列的改变, p.Ile190Asnfs* 18, 即导致第190位及以后的233个氨基酸被17个氨基酸替代, 造成编码PAX6蛋白的截短。突变前后氨基酸序列改变见图6。在家系健康成员及100名对照组健康人群中未发现该突变。
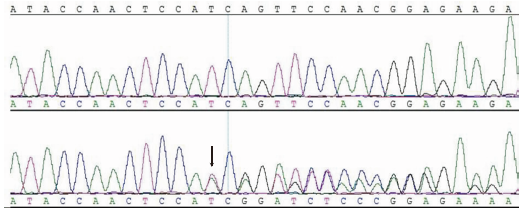 | 图5 先天性无虹膜家系患者PAX6基因第8外显子的测序突变区(c.569_570delinsACGG)。上排为正常序列, 下排为突变序列, 箭头所示为突变起始点, 根据人类基因组变异协会命名Figure 5 Sequence chromatography of the mutation identified in exon 8 of the PAX6 gene. Top panel, normal sequence; Bottom panel, mutation sequence. The initiation site of the mutation is indicated by the black arrow and was named according to the nomenclature recommended by the human gerome variation association. |
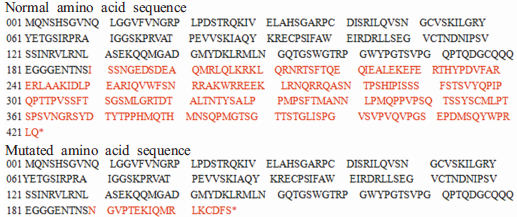 | 图6 先天性无虹膜家系患者上排位PAX6基因正常氨基酸序列, 红色为将被突变替代的233个氨基酸残基。下排为突变后的氨基酸序列, 红色为突变致蛋白截短后的17个氨基酸残基Figure 6 Amino acid sequence of PAX6. The normal amino acid sequence is shown in the top panel. The 233 amino acid residues marked in red were replaced by the 17 amino acid residues marked in red in the bottom panel. |
通过对该先天性无虹膜家系进行研究, 我们在3位患者的PAX6基因上发现一个新的杂和突变, 即2个碱基的缺失和4个碱基的插入, 造成PAX6基因编码蛋白的截短[c.569_570delinsACGG(p.Ile190Asnfs* 18)], 而在家系正常人及健康对照人群中未发现该突变。该家系3位先天性无虹膜患者的共同表现除了全部无虹膜外, 还合并有角膜病变、白内障、眼球震颤、轻度上睑下垂和黄斑发育异常等。
据研究, PAX6基因在很大程度上能调控眼球组织的生长发育, 在发育中的眼球, 视盘、视泡、视网膜、晶状体和角膜中都有PAX6基因的表达。此外, PAX6基因还在端脑、丘脑、垂体后叶素、松果体、小脑、脊髓、胰腺的发育过程中均有表达[10, 11]。目前已经确定先天性无虹膜可能与PAX6基因突变有关[12]。在这些致病突变中, 无义突变占38.9%, 剪切突变占13.2%, 框移突变占25.3%, 框内插入和缺失占6.2%, 错义突变占11.7%, 连续突变占4.7%[3]。前3种突变均导致开放的阅读框架中编码提前终止(Premature termination codon, PTC), 这导致蛋白缺失或产生截短的蛋白产物, 这种蛋白的截短是由无义介导的RNA衰减(Nonsense-mediated mRNA decay, NMD) 造成的, 从而产生无效等位基因, 其结果使PAX6基因单倍剂量不足, 造成眼部组织发育异常, 最常表现为先天性无虹膜[13]。我们在先天性无虹膜的患者中发现PAX6基因突变(c.569_570delinsACGG), 经软件预测, 该突变可产生截短蛋白p.Ile190Asnfs* 18。可以推测该突变与先天性无虹膜的致病性有关。
目前被人类基因突变数据库(HGMD)收录的PAX6基因的致病突变位点有460个(http://www.hgmd.cf.ac.uk/ac/gene.php?gene=PAX6)。突变主要导致无虹膜和其他眼部表型异常如Peters异常、中心凹发育不全和视神经发育异常等[14]。同时存在碱基的插入和缺失的报道共19个, 分别位于4-7、12外显子及第4、8、12内含子区。碱基插入和缺失突变同时存在有较高的几率发生严重的眼部症状(LOVD, http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6)。此前中国人群中只报道过一例碱基的缺失和插入同时存在[15]。张秀梅等[16]报道1个因PAX6基因第7外显子和内含子间的移码突变导致的先天性无虹膜合并上睑下垂、白内障、眼球震颤、青光眼和玻璃体混浊的家系。本研究中的先天性无虹膜患者都合并有复杂的眼部表型:轻微的眼球震颤, 轻度的上睑下垂, 不同程度的白内障, 不同程度的角膜病变和黄斑发育不良。尽管Ⅱ -3和Ⅲ -2没能完成黄斑OCT检查, 但是Ⅱ -3自幼眼球震颤, Ⅲ -2合并有高度近视和眼球震颤, 可以推测黄斑应该是发育不良的。本研究对PAX6基因直接测序, 在第8外显子上发现了新的同时存在的碱基插入和缺失突变(c.569_570delinsACGG)。
先天性无虹膜可合并有很多表型异常, 同一突变可有不同表型。即使同一家族成员的表型也可能各不相同, 而且同一个体的2只眼睛的表型也会略有不同, 对于这种由同一种致病突变导致不同表型的机制目前仍不清楚[8, 17]。本研究中的先天性无虹膜家系也验证了相同突变可致不同表型的现象。本研究中3位患者的角膜混浊程度各不相同, 白内障混浊程度也不相同, Ⅲ -2合并有高度近视, 但白内障的严重程度却比妹妹Ⅲ -3轻, 而Ⅱ -3和Ⅲ -3没有高度近视的表现。这些表型的差异表明除PAX6基因突变以外还存在其他因素, 比如表达中存在着随机变化, 而单倍体剂量不足对这种随机变化的波动又非常敏感, 因而导致疾病表型的变化[18]。
PAX6蛋白5′ 端包含2个DNA结合域, 128个氨基酸组成的配对域(Paired domain, PD)和61个氨基酸组成的同源域(Homeodomain, HD), 这2个结合域被79个氨基酸组成的连接区分开(Linker region, LNK)[19]。3′ 端是一富含脯氨酸、丝氨酸和苏氨酸的结构域, 具有转录反式激活功能[20]。本研究发现PAX6基因第8外显子的杂和突变(c.569_570delinsACGG), 经软件预测, 会导致第189位以后的233个氨基酸截短为17个氨基酸残基, 产生截短蛋白p.Ile190Asnfs* 18。该突变起始于第8外显子的LNK, 造成部分LNK及后面的HD和含脯氨酸丝氨酸和苏氨酸的结构域的蛋白截短。推测这种蛋白结构上的改变可能是该家系先天性无虹膜的致病原因。
综上所述, 本研究在我国一个先天性无虹膜家系中证实了PAX6基因第8外显子的杂合突变[c.569_570delinsACGG(p.Ile190Asnfs* 18)], 该突变在国内外尚属首次报道。本研究在分子水平上对先天性无虹膜的发病原因进行检测, 为该疾病的遗传咨询、产前诊断、基因治疗提供一定的理论依据, 也扩大了PAX6基因的突变频谱。
利益冲突申明 本研究无任何利益冲突
作者贡献声明 邢怡桥:参与选题、设计及资料的分析和解释, 对编辑部的修改意见进行修改。李印:收集数据, 撰写论文, 对编辑部的修改意见进行修改。李拓:参与选题、资料的分析和解释, 修改论文的结果、结论。李家璋:收集数据, 资料的分析和解释论文的结果、结论。杨琳:实验具体操作, 资料的分析, 撰写论文
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|